Aspectos en la implementación de consultas de alto riesgo en cáncer de páncreas
de la Morena López F, Prados Leira S
Unidad de Aparato Digestivo-Endoscopias. Hospital Universitario Madrid-Norte Sanchinarro.
Trabajo Enviado: 6/02/2015
Aceptado para su publicación: 7/03/2015
INTRODUCCIÓN
El cáncer de páncreas (CP) es una enfermedad poco conocida en relación a su etiopatogenia y de pronóstico infausto, con una supervivencia inferior al 5% a los 5 años. Este hecho se relaciona principalmente con el diagnóstico en fases avanzadas de la enfermedad, siendo del 80-90% de los pacientes diagnosticados en estadíos irresecables de inicio.
Las lesiones precursoras del CP están siendo progresivamente mejor estudiadas y comprendidas, mejorando el tratamiento y el abordaje de estos tumores en fase precoz y evitando su desarrollo. Además de las lesiones quísticas mucinosas, las neoplasias pancreáticas intraepiteliales (panIN) han suscitado el interés de los investigadores al tratarse de lesiones potencialmente precursoras del CP. La panIN-2 (displasia de bajo grado) y pan-IN3 (alto grado, carcinoma in situ) resultan de especial relevancia. La ultrasonografía endoscópica (USE) se constituye como el método de elección para el estudio pancreático dada la posibilidad de obtener material para estudio patológico y la posibilidad de estudio de cambios mínimos en el parénquima. La cirugía en estadíos precoces del CP (T1; lesiones < 20 mm) conlleva una resecabilidad del 99% con unas tasas de supervivencia, en caso de que la lesión finalmente estuviera confinada al páncreas tras estudio histopatológico (37% del total), superiores al 35% a los 5 años (1).
Los procesos neoproliferativos pancreáticos se pueden dividir en dos categorías: endocrinos y exocrinos, siendo estos últimos los más frecuentes (90-95%). EL CP exocrino (adenocarcinoma, tumor mucinoso papilar intraductal, tumor sólido pseudopapilar y carcinoma acinar) es la cuarta causa de muerte por cáncer en Estados Unidos, con un aumento de la incidencia progresiva en la última década. Un mayor estudio y conocimiento de la enfermedad ha permitido la observación de agrupaciones ocasionales de CP en determinadas familias, lo que ha “abierto la puerta” al estudio de los posibles condicionantes genéticos de dicha enfermedad. Así, aunque la mayor parte se consideren esporádicos, se estima que entre el 5-10% de los casos de CP forman parte de algún síndrome familiar.
Podemos dividir la agrupación familiar del CP en tres grupos de enfermos: familias que desarrollan CP en el seno de algún síndrome genético conocido con predisposición a malignidad pancreática, familias con pancreatitis hereditaria y por último familias con (quitar este con) dónde múltiples miembros desarrollan CP sin asociación con ningún síndrome conocido (Tabla I). Otras enfermedades poligénicas con menor impacto en el desarrollo de CP y consideradas como factores de riesgo para el desarrollo son la diabetes, obesidad y pancreatitis crónica serán también discutidas. La detección y definición del paciente en alto riesgo será un objetivo fundamental de la consulta monográfica para establecer sobre él las actividades preventivas y de vigilancia oportunas con el fin de reducir su riesgo individual y detectar en fases iniciales las lesiones con potencial curativo.
Síndromes genéticos asociados a cáncer de páncreas
| Síndrome | Mutaciones | Riesgo relativo CP |
| Peutz-Jeghers | STK11/LKB1 | 36- |
| Lynch | MLH1, MSH2 | Incrementado pero <5 |
| FAMM | CDKN2 | 16- |
| MEN-1 | MENIN | >10 |
| Síndrome mama-ovario | BRCA2 | 10- |
| Pancreatitis hereditaria | PRSS1, SPINK1 | >50 |
| P hereditario | – | 18-57 en función de nº afectos |
Tabla I
SÍNDROMES FAMILIARES Y CÁNCER DE PÁNCREAS
-
NEOPLASIAS PANCREÁTICAS ENDOCRINAS HEREDITARIAS
Estas lesiones se pueden presentar de forma esporádica o en asociación con síndromes genéticos. En general presentan una baja incidencia, la cual se estima entre 1-5 casos por millón de habitantes al año. Poco se conoce sobre las posibles bases genéticas de las lesiones esporádicas pero sí en las de componente hereditario. Principalmente la neoplasia endocrina múltiple (MEN-1) y en menor grado la esclerosis tuberosa (hamartomatosis) y el Von Hippel Lindau (carcinoma renal, angiomas en el sistema nervioso central, feocromocitoma y quistes y/o adenomas en hígado, páncreas, riñón) son factores de riesgo genético para el desarrollo de este tipo de tumores.
La MEN-1 es una rara enfermedad autonómico dominante caracterizada por la formación de múltiples tumores primarios endocrinos paratiroideos, hipofisarios y pancreáticos. La alteración de la menina producida por la mutación en dicho gen se traduce en una reducción de la capacidad supresora transcripcional de la misma con la capacidad oncogenética descrita. La edad de inicio de la afectación revela una penetrancia edad-dependiente del 45% a los 30 años, del 82% a los 50 años y del 96% a los 70 años, lo que incide en la importancia de la detección precoz y tratamiento. Existen test genéticos con una capacidad diagnóstica de hasta el 50% de las mutaciones relacionadas (2). Desde el punto de vista de los tumores endocrinos pancreáticos se ha de establecer en pacientes portadores o familiares de primer grado afectos de MEN-1 cribado mediante exploraciones de imagen pancreática bianual a partir de los 15 años de edad dada su elevada penetrancia.
-
NEOPLASIAS PANCREÁTICAS EXOCRINAS HEREDITARIAS (Tabla I)
1. Peutz-Jeghers
Síndrome autosómico dominante derivado en el 50% de los casos de la mutación de la serina-treonina kinasa (LKB1/STK11), la cual está implicada en la proliferación y polarización celular y en la vía mTOR. El efecto neto de dicha acción es la supresión tumoral que en estado mutacional pierde su capacidad.
Clínicamente cursa con la aparición de hiperpigmentación mucocutánea y pólipos hamartomatosos gastrointestinales múltiples. Se encuentra así aumentada la probabilidad de carcinoma esófago-gástrico, pulmón, intestino delgado, mama, útero, ovario y CP. Desde el punto de vista pancreático los pacientes presentan un aumento de riesgo de hasta 132 veces para el desarrollo de CP, con un riesgo acumulado a los 64 años del 36% (3). Otra lesión pancreática especialmente frecuente en este grupo de enfermos es el tumor mucinoso papilar intraductal (TPMI). De hecho se encuentran mutaciones de LKB1/STK11 en el 32% de los TMPI de la población general sin Peutz-Jeghers asociado, lo que relaciona causalmente ambos hechos.
2. Síndrome de Lynch
Síndrome autonómico dominante del que se deriva una elevada predisposición para cáncer colorectal. Se han implicado varias mutaciones de genes reparadores del ADN (hMLH1, hMSH2, hMSH6, hPMS2), que ofrecen como resultado una inestabilidad de microsatélites con un riesgo superior al 80% de padecer cáncer de colon proximal. Basado en este perfil se pueden distinguir dos subtipos sindrómicos: el tipo I sólo presenta predisposición a la neoplasia colorrectal; en el tipo II aparecen otros tumores asociados como ovario, gástrico, uterino, pancreático, biliar y urinario.
3. Síndrome de melanoma familiar atípico con
molas múltiples (FAMM)
Caracterizado por la presencia de nevus displásicos en asociación con otras malignidades como el CP. Causado por la mutación en el p16/CDKN2a localizado en el cromosoma 9p tiene una herencia de tipo autonómico dominante y alta penetrancia, en torno a un 80-100%. Las mutaciones del p16 se encuentran en más del 90% de los CP esporádicos, lo que relaciona causalmente ambas circunstancias. Se estima un riesgo relativo de 12-38 veces para CP y un riesgo acumulado del 17% a los 75 años (2). Se ha de establecer un elevado índice de sospecha de mutación del CDKN2a en pacientes con CP e historia familiar de melanoma.
4. Síndrome de cáncer hereditario mama/ovario
La relación entre las mutaciones BRCA1/BRCA2 con el aumento de riesgo de cáncer de mama y ovario está bien establecida. Las mutaciones en dichos genes presentan un patrón autonómico dominante con penetrancia incompleta, realizando un efecto supresor oncogénico. Se estima que del 5-10% de tumores de ovario se relacionan con las mutaciones del BRCA1/2. A su vez es bien conocido como las mutaciones del BRCA2 (13q) se relacionan con la aparición de CP, siendo el tercer tumor más frecuente en familias portadoras de dicha mutación. De hecho, no es infrecuente la aparición de dicha mutación en pacientes sin historia familiar de cáncer de mama, lo que confiere al BRCA2 y el CP una entidad propia. Globalmente se estima un riesgo relativo 10 veces mayor que en la población general (4).
5. Pancreatitis Hereditaria
Las mutaciones que se asocian a este síndrome clínicamente se manifiestan por el desarrollo precoz de episodios de pancreatitis aguda de tipo idiopático. Se han relacionado mutaciones de las enzimas pancreáticas con activación precoz intraglandular que origina cuadros de pancreatitis subclínica desde los 20 años de edad (PRSS1, SPINK1). Otras proteínas no exclusivas del páncreas como el intercambiador de cloro de membrana (CFTR) promueven la de la fibrosis quística (5). Por otro lado la pancreatitis tropical, propia de regiones como India y África subsahariana y también de base genética pero independiente de las anteriores, presenta características clínicas similares pero con la formación de cálculos intraductales de gran tamaño; en esta entidad se generan ya en la adolescencia de datos de insuficiencia pancreática mixta y aumenta el riesgo de cáncer pancreático al 5-100 (6).
• PRSS1 (7q35) codifica para el tripsinógeno, una serin-proteasa normalmente producida y secretada en el páncreas en forma inactiva hasta su activación en el intestino delgado. Es la alteración genética más frecuente, en torno al 70% de los casos. La mutación sobre PRSS1, hereditaria de manera autosómica dominante (no coma) en sus formas más frecuentes (R122H y N29I) traduce la activación de la enzima intraglandular, lo que produce la autodigestión pancreática y el fenómeno inflamatorio secundario. Así y a diferencia de las anteriores, el efecto mutacional no se evidencia en una alteración oncogénica determinada sino que el efecto neoplásico se deriva de los sucesivos fenómenos de inflamación, regeneración y fibrosis de las sucesivas pancreatitis. El riesgo relativo de CP a los 70 años se estima entre el 53-70% de los pacientes afectos por esta mutación. En caso de tabaquismo activo la cifra se eleva hasta las 150 veces.
• SPINK1 y sus variantes denotan una herencia autosómico recesiva con penetrancia variable, por lo que raramente las familias de los pacientes afectos presentan historia de pancreatitis de repetición. El efecto neto de la alteración es un disbalance entre las proteasas y sus inhibidores a nivel intrapancreático, con la activación prematura (nuevamente) de la tripsina con los mismos efectos que PRSS1. El riesgo de CP se encuentra también aumentado en esta variante.
• Las mutaciones sobre el gen de la fibrosis quística (CFTR) se asocian también a formas precoces e idiopáticas de pancreatitis aguda. Estas mutaciones se encuentran hasta en el 5% de la población y serían las responsables de hasta el 50% de pancreatitis agudas idiopáticas. De hecho, y aunque para el desarrollo de fibrosis quística por su herencia recesiva sea preciso la unión de dos portadores por su herencia recesiva, para la manifestación de pancreatitis aguda recurrente sólo precisa de una única mutación (7).
6. Cáncer pancreático familiar
La agregación de casos de CP en familias se encuentra en alrededor del 10% de todos los casos. La ausencia de síndrome genético conocido y la aparición en la misma familia de al menos dos afectos de primer grado determinan el síndrome conocido como cáncer pancreático familiar. Así, el riesgo de un individuo con dos familiares de primer grado afectados por CP eleva su probabilidad individual hasta las 18 veces, aumentando de 32 a 45 si hay tres ó más (Tabla II) (8), no negrita. Aunque las bases genéticas de esta forma sigan siendo desconocidas se han realizado avances en el ámbito genético; la presencia de mutación BRCA2 se ha evidenciado en el 6-17% de estas familias.
Riesgo de desarrollo de cp en formas de cáncer pancreático familiar
| Número de familiares afectos | Incremento de Riesgo Relativo |
| 1 | 4-5 veces |
| 2 | 18 veces |
| >3 | 2 a 57 veces |
Tabla II
III. OTROS FACTORES DE RIESGO
1.- Diabetes mellitus
Metanálisis recientes demuestran que la presencia de diabetes aumenta en 2,1 veces el riesgo relativo de padecer un CP, especialmente tras los 5 primeros años tras el diagnóstico (9). Aún cuando dicha asociación exista no se puede considerar la diabetes de forma aislada como un factor con un peso relevante para determinar de forma individual un paciente como en riesgo de CP. Sin embargo el CP sí se relaciona de forma importante con la producción de intolerancia a los hidratos de carbono; la diabetes acontece entre el 50-65% de CP, ocurriendo con una asociación temporal muy próxima al inicio de la actividad tumoral. Así, pacientes con debut diabético por encima de los 50 años de edad presentan un riesgo individual para CP de hasta 10 veces en los tres primeros años tras el diagnóstico. Por todo ello se postula la intolerancia a los hidratos de carbono como un posible test de screening para el CP resecable.
2.- Obesidad
De forma aislada no conforma un elemento de peso relevante para el desarrollo de CP pero sí un aumento discreto del riesgo. Pacientes con un índice de masa corporal (IMC) >30 presentan un aumento de riesgo de 1,81 veces respecto a IMC <25, con una estratificación por sexos en varones con más de 35 de IMC de 2,61 veces y mujeres con más de 40 de IMC de 2,76 (10).
3.- Pancreatitis crónica
Los pacientes con pancreatitis crónica presentan un riesgo relativo individual de 14,4-16,5 veces más de riesgo para CP, siendo la pancreatitis crónica secundaria a pancreatitis hereditaria recurrente un factor de riesgo separado y mucho más importante en su potencial degenerativo que el resto de causas. Así, y aunque sólo 1-2% de los pacientes con pancreatitis crónica generen un CP, la incidencia acumulada de lesiones malignas se sitúa en torno al 2% por década, estando muy relacionado con la edad (3 veces más en pacientes de 40-60 años y hasta 10 veces más en pacientes de 60 años) y con el consumo de alcohol y tabaco, que duplican todos los datos anteriores en caso de consumo activo (11).
4.- Miscelanea
Ataxia-Telangiectasia, Anemia de Fanconi, Poliposis Adenomatosa familiar y Li-Fraumeni
Son factores de escasa relevancia, con bajo impacto en el riesgo individual o relacionados pero de riesgo desconocido.
CRIBADO Y MÉTODOS DIAGNÓSTICOS
El objetivo último de las estrategias de cribado y vigilancia ha de ser el caracterizar lesiones precoces y sin capacidad de invasión que las definan como no resecables. Las lesiones precursoras de neoplasia pancreática son las quísticas mucinosas (caracterizadas en otro protocolo) y las neoplasias pancreáticas intraepiteliales o PanINs. Éstas últimas tienen un interés especial por su difícil diagnóstico e importancia como fases iniciales de la mayor parte de adenocarcinomas pancreáticos. Las PanINs se componen generalmente de epitelio columnar y cuboidal con varios grados de atipia, afectando a los ductos pancreáticos pequeños y (quitaría el que) son indetectables en las pruebas de imagen convencionales. Las PanIN1 son hiperplásicas con un mínimo grado de atipia y pueden subdividirse en tipo A ó B en función de de la presencia o ausencia de crecimientos micropapilares respectivamente. Por otro lado las PanIN2 son lesiones con displasia de bajo grado e incluyen cambios celulares, pérdida de polaridad, pseudoestratificación, hipercromatismo y núcleos alargados. Por último las PanIN3 son lesiones de alto grado o carcinoma in situ, con la presencia de mitosis atípicas, necrosis luminal pero contenidas por la membrana basal (12).
Sensibilidad y especificidad de los estudios de imagen y diagnóstico molecular en cp
Global CP | CP< 2cm | CP<1cm | ||
| Test | Sensibilidad | Especificidad | Sensibilidad | Especificidad |
| Ecografía abdominal | 56-97 | 50-90 | 65 | 23 |
| TAC abdominal | 53-91 | 53-93 | 40-65 | 15 |
| RMN | 83-87 | 81-100 | 33 | – |
| CPRE | 70-97 | 81-100 | 93 | 91 |
| Ecoendoscopia | 86-100 | 69-100 | 74-90 | – |
| p53 en líquido pancreático | 42-67 | 59-100 | – | – |
| k-ras en líquido pancreático | 63-100 | 50-100 | – | – |
Tabla III
Genéticamente se identifica la presencia de mutaciones como k-ras, p16 y alargamiento telomérico en un gran número de estas lesiones. La detección de k-ras mutado en el jugo duodenal o pancreático aspirado tras estimulación con secretina es muy sensible aunque poco específico, particularmente en pacientes con pancreatitis crónica. La presencia de p53, CDKN2A y SMAD4 resulta menos sensible pero más específica para displasia. Se ha propuesto una estrategia escalonada (EUROPAC) basada en los resultados del aspirado mediante CPRE de fluido pancreático con la detección secuencial, en caso de que k-ras fuese positivo, de los otros marcadores como valores de detección precoz de CP inicial (13). Se sugiere que estas lesiones, al encontrarse en comunicación con el sistema ductal pancreático, pueden promover niveles elevados de dichos marcadores en fluido pancreático, lo cual demuestra utilidad como potencial método de diagnóstico precoz.
Estratificación pacientes en función del riesgo genético
| Bajo riesgo (<5) | Riesgo moderado (5-10) | Alto riesgo (>10) |
| Un familiar CP Mutantes portadores BRCA2 Pancreatitis hereditaria | Mutantes portadores BRCA2 | Pancreatitis hereditaria |
| Lynch | Pacientes con fibrosis quística | Peutz-Jeghers |
| Mutantes portadores BRCA1 | Pacientes con pancreatitis crónica | MEN-1 |
| Poliposis adenomatosa familiar | Un familiar de primer grado con CP menor de 55 años Dos familiares con CP independientemente del grado | Tres o más familiares independientemente del grado con CP Dos familiares con CP, uno de primer grado y otro de segundo siendo uno menor de 55 años Dos o más familiares de primer grado CP FAMM |
Tabla IV
Aunque no se produzcan cambios patognomónicos en el parénquima pancreático la ecoendoscopia es el único método definido como útil para la valoración de estas lesiones, siendo la mejor prueba diagnóstica para lesiones neoplásicas subcentimétricas en el cual se englobarían las PanIN. Aunque los cambios producidos por los PanIN sean superponibles a los de la pancreatitis crónica, la presencia de micromódulos hipoecoicos se ha sugerido como el factor más correlacionable con estas lesiones sobre otros (lo quitaría) (14). Adicionalmente la ecoendoscopia aporta el teórico potencial diagnóstico de la PAAF, aunque su rentabilidad en el seno de la detección de las PanIN2-3 no ha sido aún contrastada. La progresión de dichas lesiones, aún en estadíos precoces, promoverá alteraciones ductales potencialmente diagnosticables mediante RMN pancreática y/o CPRE, dejando ésta última opción por su carácter invasivo como método para el estudio de marcadores en el fluido pancreático (Tabla III) (15)
Con el fin de estratificar correctamente a los pacientes en función de su riesgo y para la optimización de los recursos en una consulta de cáncer pancreático familiar se han establecido tres grupos de enfermos. Se consideran de bajo riesgo aquellos con un riesgo acumulado menor de 5, (quitado los) de riesgo intermedio si es de 5-10 y con riesgo alto si es mayor de 10 (Tabla IV). (16)
Se establece adecuada la indicación de cribado y vigilancia a todos los pacientes con riesgo alto y moderado, siendo sólo incluidos en la consulta aquellos pacientes con riesgo bajo que así lo soliciten.
La edad adecuada para iniciar el cribado no ha sido establecida, por lo que se suele extrapolar de consideraciones relacionadas con el cáncer colorectal. Así, se recomienda su inicio a partir de los 50 años ó 10 años antes del familiar afecto más joven de la estirpe. Sin embargo se han encontrado familias afectas con cierto componente de anticipación genética en las sucesivas herencias por lo que el inicio puede incluso adelantarse más en la edad, especialmente y hasta 10 años más en pacientes fumadores. La únicas indicaciones claras al respecto las conforman los pacientes con MEN-1, con indicación de exploración pancreática bianual a partir de los 15 años de edad, Peutz-Jeghers a partir de los 25-30 años y pancreatitis hereditaria con una frecuencia anual a partir de 40 años. En relación al resto de síndromes la frecuencia de las exploraciones se recomienda bianual o incluso cada 3 años hasta la edad del primer familiar afecto y anual a partir de la misma (17).
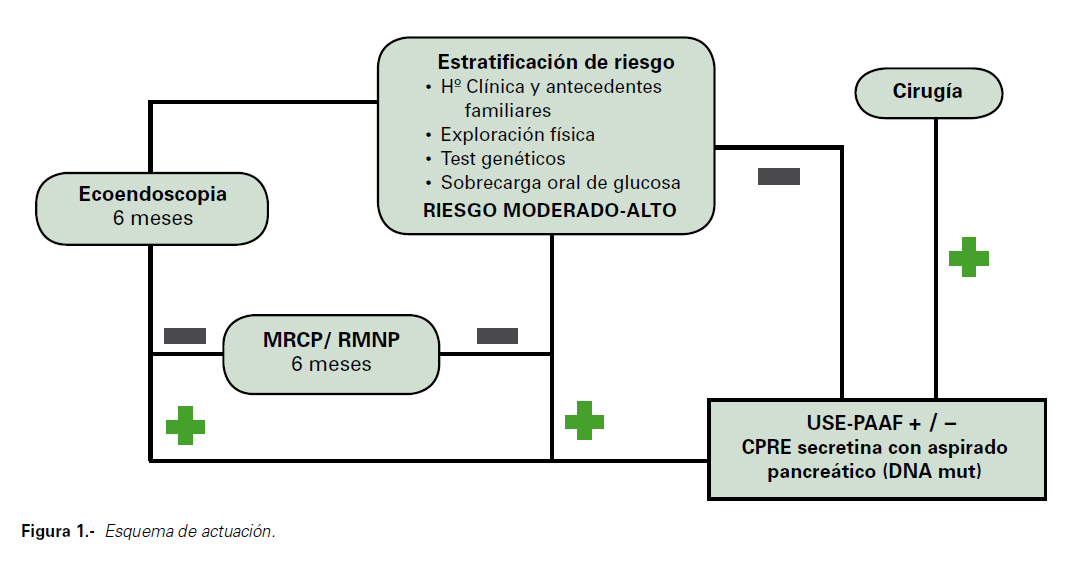
Se propone un esquema de actuación (Figura 1).

ACTUACIONES TERAPÉUTICAS
-
ACTUACIONES ENCAMINADAS A LA REDUCCIÓN DE RIESGO INDIVIDUAL
1.- Dieta y ejercicio
Se ha de recomendar dieta y ejercicio físico regular a todos los familiares ya que en estudios epidemiológicos la ingesta de grasa, exceso de calorías e hidratos de carbono se ha asociado con un riesgo aumentado de CP; las dietas ricas en fruta y verduras lo reducen poblacionalmente. De igual forma, las poblaciones que realizan semanalmente 1,5 horas de ejercicio físico suave presentan un 50% menos de incidencia de CP que aquellas totalmente sedentarias (18).
2.- Ingesta de alcohol
La eliminación de este factor de riesgo sólo previene la aparición de CP si se suprime previamente a la formación de pancreatitis crónica. Como se comentó anteriormente, la pancreatitis crónica es un factor de riesgo independiente para CP.
3.- Tabaquismo
Es un factor clave y claramente relacionado con el CP, aumentando el riesgo en 2-3 veces en la población general pero duplicándolo y haciendo su aparición más precoz en la población de riesgo. Se considera que si se produjera el cese completo del tabaquismo a nivel mundial la prevalencia de CP caería en un 22% (19).
4.- Tratamiento farmacológico
Se ha especulado sobre cómo el uso de AINE´s podría reducir el riesgo teórico de CP. Estudios epidemiológicos no verifican dicha hipótesis por lo que no se recomienda su uso como quimioprevención.
-
TRATAMIENTO QUIRÚRGICO
Una vez que la lesión displásica ha sido diagnosticada en un paciente de alto riesgo el mejor tratamiento disponible es la resección quirúrgica en términos de reducción en la mortalidad por CP. Sin embargo el carácter multifocal de los PanIN convierte la extensión de la cirugía pancreática en un punto controvertido. El hallazgo de lesiones tipo PanIN en los fragmentos resecados infiere un carácter multifocal hasta en el 73% de los pacientes en algunas series. Por ello son aceptables tres tipos de actitudes a consensuar con el paciente en función de su riesgo y características, ya que no existe hoy en día actitud de consenso clara al respecto:
a) Pancreatectomía total inicial.
b) Pancreatectomía distal y en caso de hallazgo de PanIN multifocal o tipo 3 proceder a completar la cirugía a total en un segundo tiempo.
c) Pancreatectomía distal y vigilancia estricta del muñón residual.
NOTA DEL AUTOR
El presente protocolo no presenta conflictos de interés y ha sido elaborado mediante búsqueda sistemática bibliográfica de revisiones y originales
BIBLIOGRAFÍA
1. Tsuchiya R, Noda T, Harada N, Miyamoto T, Tomioka T, Yamamoto K, et al. Collective review of small carcinomas of the pancreas. Annals of Surgery 1986;203(1):77-81.
2. Nagy R, Sweet K, Eng C. Highly penetrant hereditary cancer syndromes. Oncogene 2004;23(38):6445-70.
3. Giardiello FM, Brensinger JD, Tersmette AC, Goodman SN, Petersen GM, Booker SV, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology 2000;119(6):1447-53.
4. Greer JB, Whitcomb DC. Role of BRCA1 and BRCA2 mutations in pancreatic cancer. Gut 2007;56(5):601-5.
5. Simon P, Weiss FU, Sahin-Toth M, Parry M, Nayler O, Lenfers B, et al. Hereditary pancreatitis caused by a novel PRSS1 mutation (Arg-122 –> Cys) that alters autoactivation and autodegradation of cationic trypsinogen. The Journal of Biological Chemistry 2002;277(7):5404-10.
6. Chari ST, Mohan V, Pitchumoni CS, Viswanathan M, Madanagopalan N, Lowenfels AB. Risk of pancreatic carcinoma in tropical calcifying pancreatitis: an epidemiologic study. Pancreas 1994;9(1):62-6.
7. Teich N, Mossner J. Hereditary chronic pancreatitis. Best Practice & Research Clinical Gastroenterology 2008;22(1):115-30.
8. Klein AP, Brune KA, Petersen GM, Goggins M, Tersmette AC, Offerhaus GJ, et al. Prospective risk of pancreatic cancer in familial pancreatic cancer kindreds. Cancer Research 2004;64(7):2634-8.
9. Eberle MA, Pfutzer R, Pogue-Geile KL, Bronner MP, Crispin D, Kimmey MB, et al. A new susceptibility locus for autosomal dominant pancreatic cancer maps to chromosome 4q32-34. American Journal of Human Genetics 2002;70(4):1044-8.
10. Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. The New England Journal of Medicine 2003;348(17):1625-38.
11. Yeo TP, Hruban RH, Brody J, Brune K, Fitzgerald S, Yeo CJ. Assessment of “gene-environment” interaction in cases of familial and sporadic pancreatic cancer. Journal of gastrointestinal surgery : official journal of the Society for Surgery of the Alimentary Tract 2009;13(8):1487-94.
12. Hruban RH, Adsay NV, Albores-Saavedra J, Compton C, Garrett ES, Goodman SN, et al. Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. The American Journal of Surgical Pathology 2001;25(5):579-86.
13. Vitone LJ, Greenhalf W, McFaul CD, Ghaneh P, Neoptolemos JP. The inherited genetics of pancreatic cancer and prospects for secondary screening. Best Practice & Research Clinical Gastroenterology 2006;20(2):253-83.
14. Kimmey MB, Bronner MP, Byrd DR, Brentnall TA. Screening and surveillance for hereditary pancreatic cancer. Gastrointestinal Endoscopy 2002;56(4 Suppl):S82-6.
15. Pongprasobchai S, Chari ST. Management of Patients at High Risk for Pancreatic Cancer. Current Treatment Options in Gastroenterology 2003;6(5):349-58.
16. Verna EC, Hwang C, Stevens PD, Rotterdam H, Stavropoulos SN, Sy CD, et al. Pancreatic cancer screening in a prospective cohort of high-risk patients: a comprehensive strategy of imaging and genetics. Clinical cancer research : an official journal of the American Association for Cancer Research 2010;16(20):5028-37.
17. Larghi A, Verna EC, Lecca PG, Costamagna G. Screening for pancreatic cancer in high-risk individuals: a call for endoscopic ultrasound. Clinical cancer research : an official journal of the American Association for Cancer Research 2009;15(6):1907-14.
18. Michaud DS, Giovannucci E, Willett WC, Colditz GA, Stampfer MJ, Fuchs CS. Physical activity, obesity, height, and the risk of pancreatic cancer. JAMA : the journal of the American Medical Association 2001;286(8):921-9.
19. Yamaguchi K. How to define patients at high risk for pancreatic cancer. Pancreatology : official journal of the International Association of Pancreatology 2011;11 Suppl 2:3-6.